




La fibrilación auricular (FA) es la arritmia más común en la práctica clínica, y afecta aproximadamente al 1-4% de la población general. Los estudios indican una prevalencia notablemente más baja de FA en las personas menores de 50 años, siendo del 1% entre aquellos de entre 50-59 años y del 0,3% entre los de 40-49 años1. Es relevante la presencia de antecedentes familiares de FA, bloqueo auriculoventricular (BAV) de inicio temprano o casos de miocardiopatía y muerte súbita cardiaca (MSC) en algunos jóvenes que experimentan el inicio de la FA2. Se ha sugerido que la herencia genética desempeña un papel fundamental en estos casos, dado que se ha asociado la FA de aparición precoz con mutaciones en genes relacionados con miocardiopatías hereditarias y síndromes arrítmicos primarios3. Yoneda et al. analizaron genéticamente a 1.293 pacientes con FA de inicio temprano (<66 años) e identificaron una variante asociada a la enfermedad en aproximadamente el 10%4. El rendimiento del estudio fue mayor en los pacientes diagnosticados antes de los 30 años (16,8%) y menor en aquellos diagnosticados después de los 60 años. Las variantes asociadas a la enfermedad fueron más comunes en genes asociados con miocardiopatías, especialmente en TTN. En concreto, en el 2,1% de todos los pacientes con FA de inicio temprano, y en el 6,5% de los pacientes diagnosticados antes de los 30 años, las mutaciones con pérdida de función en TTN fue el sustrato genético identificado3.
No existen recomendaciones robustas para la realización de un estudio genético en pacientes con FA a edades tempranas. Las recientes guías de FA de la ACC/AHA/ACCP/HRS del año 2023 sugieren que el estudio genético podría ser razonable (2b; B-NR) en los pacientes <45 años sin factores de riesgo obvios de FA5. El presente trabajo pretende describir el rendimiento del estudio genético en vida real en los pacientes con inicio de FA a edades tempranas.
Se trata de un estudio observacional retrospectivo multicéntrico de los pacientes estudiados en consulta de cardiopatías familiares de 3 hospitales, cuyo evento clínico índice fue la FA, y que cumplían alguna de las siguientes condiciones: a) Primer episodio de FA antes de los 35 años; b) Pacientes con primer episodio de FA antes de los 50 años, sin diagnóstico previo de miocardiopatía que presentaran bien algún grado de cardiopatía estructural (diámetro telediastólico del ventrículo izquierdo [VI]≥55mm, hipertrofia del VI>12mm o fracción de eyección del VI [FEVI]<50%) o bien antecedentes familiares antes de los 50 años de FA, MSC inexplicada o bloqueo auriculo ventricular avanzado, y c) Primer episodio de FA antes de los 50 años en los pacientes incluidos en estudio en cascada por otra cardiopatía familiar. A todos ellos se les solicitó estudio genético.
Se incluyeron a los pacientes cuyo estudio genético se realizó entre enero de 2019 y diciembre de 2023, mediante paneles de NGS comerciales (Health in Code, España) dirigido al fenotipo índice en todos los casos (FA, 55 genes; miocardiopatía hipertrófica, 140 genes; miocardiopatía dilatada, 146 genes; MSC, 288 genes; arritmias y MSC sin cardiopatía estructural, 100 genes), excepto en los identificados mediante cribado en cascada. Se consideraron portadores de mutaciones (G+) las variantes genéticas clasificadas como patogénicas o posiblemente patogénicas según la clasificación del American College of Medical Genetics. Se recogió información acerca de los antecedentes familiares y la historia clínica. El trabajo se realizó de acuerdo con los principios éticos de la Declaración de Helsinki, se recogió consentimiento informado de los pacientes y contó con la aprobación del Comité de Ética provincial de Almería.
Se incluyó a 25 pacientes (edad media=37,4±10 años; 7 [28%] mujeres). En el 48% de los casos se trataba de FA paroxística, el resto se presentó como FA persistente. El CHADS2VASC2 medio fue de 1,08±1,07. No se comunicó consumo de alcohol excesivo regular ni de otros tóxicos. La cohorte presentó una FEVI media de 55±12% y un diámetro telediastólico del VI medio de 49,9±9mm. Hubo 9 pacientes con FEVI reducida (42±13%) y dilatación leve del VI (diámetro telediastólico del VI, 59,1±5mm). Cuatro pacientes reunían criterios de miocardiopatía hipertrófica. El 48% se sometió al menos a un procedimiento de ablación de venas pulmonares, y 9 pacientes (36%) se mantuvieron en FA permanente.
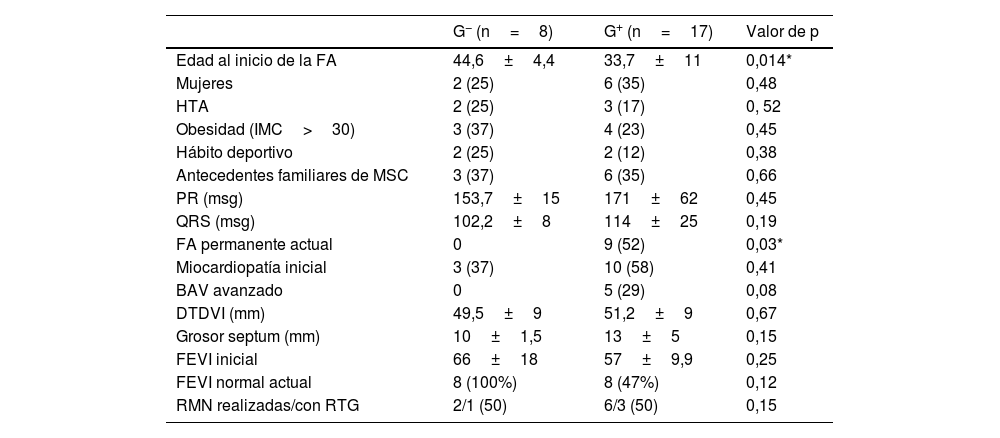
Se realizaron 21 paneles de NGS (3 de FA, 4 de miocardiopatía hipertrófica, 9 paneles de miocardiopatía dilatada, 4 paneles MSC y un panel de canalopatías). En otros 4 casos se realizó secuenciación puntual. El estudio genético fue positivo (G+) en 17 casos (68%). De los 17 (G+), 14 eran probandos, con panel NGS dirigido a fenotipo, el resto se identificó mediante el estudio en cascada. Los genes más frecuentemente implicados fueron LMNA (6 pacientes, 5 mutaciones distintas) y TTN (5 pacientes, 4 mutaciones). En un paciente se halló una mutación en un gen codificante de canal de sodio (SCN5A). La frecuencia y tipo de las mutaciones están recogidas en la figura 1. Los pacientes G+ fueron significativamente más jóvenes y desarrollaron más FA permanente. Aunque no fue estadísticamente significativo, se observó en el grupo G+ la presencia de más mujeres. Se observó mayor tendencia a que presentaran antecedentes familiares de MSC, desarrollaran más BAV avanzado en su evolución y presentaran una FEVI normal, aunque menor que el grupo G−. La tabla 1 resume estos hallazgos.
: resultado de panel genético negativo; LMNA: mutación en gen de la lamina; MYH7: mutación en gen MYH7(myosin heavy chain 7); PRKAG2: mutación en gen PRKAG2(protein kinase AMP-activated non-catalytic subunit gamma 2); SCN5A: mutación en gen del canal de sodio subunidad 5 alfa; TNNI3: mutación en gen de la troponina I3; TTN: mutación en gen de la titina.")
Frecuencias de resultados genéticos. Genotipo (−): resultado de panel genético negativo; LMNA: mutación en gen de la lamina; MYH7: mutación en gen MYH7(myosin heavy chain 7); PRKAG2: mutación en gen PRKAG2(protein kinase AMP-activated non-catalytic subunit gamma 2); SCN5A: mutación en gen del canal de sodio subunidad 5 alfa; TNNI3: mutación en gen de la troponina I3; TTN: mutación en gen de la titina.
Características de la población con estudio genético negativo (G−) frente al estudio genético positivo (G+)
| G− (n=8) | G+ (n=17) | Valor de p | |
|---|---|---|---|
| Edad al inicio de la FA | 44,6±4,4 | 33,7±11 | 0,014* |
| Mujeres | 2 (25) | 6 (35) | 0,48 |
| HTA | 2 (25) | 3 (17) | 0, 52 |
| Obesidad (IMC>30) | 3 (37) | 4 (23) | 0,45 |
| Hábito deportivo | 2 (25) | 2 (12) | 0,38 |
| Antecedentes familiares de MSC | 3 (37) | 6 (35) | 0,66 |
| PR (msg) | 153,7±15 | 171±62 | 0,45 |
| QRS (msg) | 102,2±8 | 114±25 | 0,19 |
| FA permanente actual | 0 | 9 (52) | 0,03* |
| Miocardiopatía inicial | 3 (37) | 10 (58) | 0,41 |
| BAV avanzado | 0 | 5 (29) | 0,08 |
| DTDVI (mm) | 49,5±9 | 51,2±9 | 0,67 |
| Grosor septum (mm) | 10±1,5 | 13±5 | 0,15 |
| FEVI inicial | 66±18 | 57±9,9 | 0,25 |
| FEVI normal actual | 8 (100%) | 8 (47%) | 0,12 |
| RMN realizadas/con RTG | 2/1 (50) | 6/3 (50) | 0,15 |
Los datos expresan n (%) o media±desviación estándar.
BAV: bloqueo auriculoventricular; DTDVI: diámetro telediastólico de ventrículo izquierdo; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; HTA: hipertensión arterial; IMC: índice de masa corporal; MSC: muerte súbita cardiaca; PR: medida intervalo PR del ECG; QRS: medida intervalo QRS del ECG; RMN: resonancia magnética nuclear; RTG: realce tardío con gadolinio.
La observación clínica de que algo más de un 60% de los pacientes presentó mutaciones relevantes, casi todas en genes responsables de miocardiopatías, como en la serie de Yoneda et al.4, subrayan la necesidad de integrar la genética en la evaluación etiológica de la FA de inicio precoz, especialmente cuando se acompaña alguna de las circunstancias referidas.
El hallazgo de hasta 5 mutaciones distintas en LMNA en 6 pacientes que se manifiestan con FA jóvenes o con antecedentes familiares de arritmias o MSC tiene implicaciones pronósticas de aplicación inmediata en el probando y familiares. Por otro lado, es conocida la prevalencia de mutaciones en TTN en miocardiopatías como la alcohólica6 o el periparto. Su prevalencia en pacientes jóvenes con FA como primera manifestación, arroja la pregunta de qué casos de taquimiopatías en pacientes jóvenes pueden obedecer a un componente genético más allá de la propia taquiarritmia.
Nuestros resultados muestran una tasa muy significativa de mutaciones genéticas en esta población muy seleccionada de pacientes jóvenes con FA de inicio. Al tratarse de un trabajo retrospectivo y en pacientes de una consulta de cardiopatías familiares, estas conclusiones están sometidas a un evidente sesgo de selección. Se han excluido pacientes diagnosticados previamente de miocardiopatía hipertrófica o miocardiopatía dilatada, o una canalopatía que presenta una FA a posteriori o como epifenómeno.
Según el estudio de Yoneda et al.4, la aplicación práctica de la genética sistemática en individuos menores de 66 años en práctica clínica diaria se ve desafiada por su modesto rendimiento diagnóstico (∼10%) y la considerable asignación de recursos requeridos. Las mencionadas guías americanas de 2023 sugieren que el estudio genético podría ser razonable en pacientes menores de 45 años sin factores de riesgo obvios de FA5. Esta indicación genérica mejoraría ligeramente su rendimiento, aunque con una precisión probablemente aún reducida. Nos planteamos la hipótesis de que acotando los casos a la población joven con inicio de FA con mayor probabilidad pre-test, como la referida en este trabajo, el estudio genético podría ser verdaderamente rentable. Cambiaría el enfoque diagnóstico y de tratamiento, especialmente en pacientes portadores de mutaciones con implicaciones pronósticas relevantes. Sin embargo, se necesitan más estudios prospectivos para validar estos hallazgos y determinar el impacto futuro de la detección temprana de mutaciones genéticas en pacientes con FA a edades precoces.
FinanciaciónNo se ha solicitado ni recibido financiación adicional para la realización del estudio.
Consideraciones éticasEl estudio fue aprobado por el Comité de Ética e Investigación Provincial de Almería, y se obtuvo el consentimiento informado de los pacientes. El trabajo se realizó de acuerdo con los principios éticos de la Declaración de Helsinki. Confirmamos que los posibles sesgos en las variables de sexo/género se han tenido en cuenta de acuerdo con las directrices SAGER.
Declaración sobre el uso de inteligencia artificialNo se ha empleado inteligencia artificial en el desarrollo de este artículo.
Contribución de los autoresTodos los autores han hecho contribuciones sustanciales en la concepción y el diseño del estudio, la adquisición de datos, el análisis y la interpretación de los datos, además de haber aprobado el borrador del artículo y haber hecho una revisión crítica del contenido intelectual.
Conflicto de interesesNo existen conflictos de intereses de los autores.



