




En el estudio PARADIGM-HF1, el sacubitrilo-valsartán (SV), inhibidor de la neprilisina y del receptor de la angiotensina, demostró superioridad frente al enalapril en la reducción de la morbimortalidad en pacientes ambulatorios con insuficiencia cardiaca (IC) con fracción de eyección reducida (ICFEr). Más recientemente, en los estudios TRANSITION2,3 y PIONEER-HF4 se observó que el SV puede iniciarse de forma segura y precoz en pacientes hospitalizados tras una descompensación por IC aguda, durante la hospitalización o precozmente tras el alta. Sin embargo, no hay datos en vida real respecto a esta indicación.
El objetivo de este estudio es evaluar la seguridad y la tolerabilidad del SV en pacientes hospitalizados por un episodio de IC descompensada (perfil clínico similar a los del estudio TRANSITION) frente a pacientes ambulatorios (perfil clínico similar a los del estudio PARADIGM-HF) en una cohorte de pacientes de la vida real.
Realizamos un subanálisis de los pacientes del registro Sacubitril/Valsartan Evidences in Real Life (SAVE-RLIFE). Se trata de un estudio observacional, ambispectivo y multidisciplinar en el que se incluyó a pacientes con ICFEr que iniciaron SV entre septiembre de 2016 y octubre de 2018. El registro (AEMPS MGR-SAC-2018-01) se realizó conforme a las normas de buena práctica clínica y fue aprobado por el comité de ética del hospital.
Se incluyó a 231 pacientes, edad media de 65,43 ± 11,80 años, 25% mujeres. De ellos, 48 pacientes (20,8%) iniciaron el tratamiento con SV durante la hospitalización tras un episodio de descompensación de IC y 183 pacientes (79,2%) lo iniciaron ambulatoriamente según las recomendaciones de las guías clínicas5. La mediana de seguimiento fue 338 días (Q1: 215; Q3: 563) en el subgrupo de pacientes ambulatorios y de 218 días (Q1: 128; Q3: 455) en los hospitalizados.
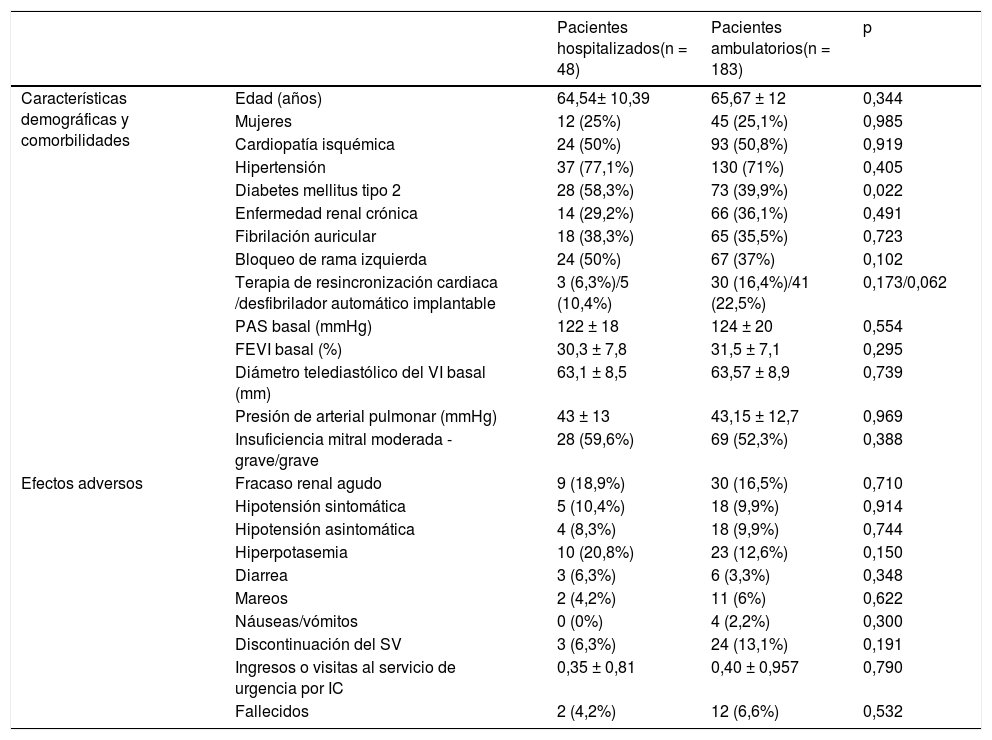
Las características demográficas y las comorbilidades basales se presentan en la tabla 1. Se observa que ambos subgrupos son homogéneos, salvo una mayor prevalencia de diabetes entre los pacientes con inicio del tratamiento hospitalizado y mayor uso de dispositivos en los pacientes ambulatorios. En las características basales, se objetivan unas cifras medias de presión arterial sistólica (PAS) al inicio del fármaco similares en ambos grupos (122 ± 18mmHg y 124 ± 20mmHg, p = 0,554, en el grupo de hospitalizados y ambulatorios respectivamente), similar a la cohorte del ensayo clínico TRANSITION2,3 (124 ± 14mmHg) y ligeramente mayor al ensayo PIONEER-HF4 (118 [110; 133] mmHg).
Características basales, seguridad y efectos adversos entre ambos subgrupos
| Pacientes hospitalizados(n = 48) | Pacientes ambulatorios(n = 183) | p | ||
|---|---|---|---|---|
| Características demográficas y comorbilidades | Edad (años) | 64,54± 10,39 | 65,67 ± 12 | 0,344 |
| Mujeres | 12 (25%) | 45 (25,1%) | 0,985 | |
| Cardiopatía isquémica | 24 (50%) | 93 (50,8%) | 0,919 | |
| Hipertensión | 37 (77,1%) | 130 (71%) | 0,405 | |
| Diabetes mellitus tipo 2 | 28 (58,3%) | 73 (39,9%) | 0,022 | |
| Enfermedad renal crónica | 14 (29,2%) | 66 (36,1%) | 0,491 | |
| Fibrilación auricular | 18 (38,3%) | 65 (35,5%) | 0,723 | |
| Bloqueo de rama izquierda | 24 (50%) | 67 (37%) | 0,102 | |
| Terapia de resincronización cardiaca /desfibrilador automático implantable | 3 (6,3%)/5 (10,4%) | 30 (16,4%)/41 (22,5%) | 0,173/0,062 | |
| PAS basal (mmHg) | 122 ± 18 | 124 ± 20 | 0,554 | |
| FEVI basal (%) | 30,3 ± 7,8 | 31,5 ± 7,1 | 0,295 | |
| Diámetro telediastólico del VI basal (mm) | 63,1 ± 8,5 | 63,57 ± 8,9 | 0,739 | |
| Presión de arterial pulmonar (mmHg) | 43 ± 13 | 43,15 ± 12,7 | 0,969 | |
| Insuficiencia mitral moderada - grave/grave | 28 (59,6%) | 69 (52,3%) | 0,388 | |
| Efectos adversos | Fracaso renal agudo | 9 (18,9%) | 30 (16,5%) | 0,710 |
| Hipotensión sintomática | 5 (10,4%) | 18 (9,9%) | 0,914 | |
| Hipotensión asintomática | 4 (8,3%) | 18 (9,9%) | 0,744 | |
| Hiperpotasemia | 10 (20,8%) | 23 (12,6%) | 0,150 | |
| Diarrea | 3 (6,3%) | 6 (3,3%) | 0,348 | |
| Mareos | 2 (4,2%) | 11 (6%) | 0,622 | |
| Náuseas/vómitos | 0 (0%) | 4 (2,2%) | 0,300 | |
| Discontinuación del SV | 3 (6,3%) | 24 (13,1%) | 0,191 | |
| Ingresos o visitas al servicio de urgencia por IC | 0,35 ± 0,81 | 0,40 ± 0,957 | 0,790 | |
| Fallecidos | 2 (4,2%) | 12 (6,6%) | 0,532 | |
FEVI: fracción de eyección del ventrículo izquierdo; IC: insuficiencia cardiaca; PAS: presión arterial sistólica; SV: sacubitrilo-valsartán; VI: ventrículo izquierdo.
Los datos se expresan en n (%) o media ± desviación estándar.
Al analizar la evolución de la clase funcional antes y después del uso del SV (fig. 1), se observa una mejoría de la clase funcional New York Heart Association (NYHA) y la ausencia de disnea en ambos subgrupos (NYHA I en el 35,7 y 27,2% en pacientes hospitalizados frente a ambulatorios, respectivamente, p = 0,281). Sin embargo, considerando la mejoría de la clase funcional, no se observa diferencia entre ambos subgrupos (59,5 durante el ingreso y 54,8% ambulatorios; p = 0,582). Dado que la única comorbilidad que difiere es la diabetes, se realizó un análisis de regresión logística, ajustado por diabetes y se observó que no hay asociación entre tener diabetes y presentar o no una mejoría de la clase funcional (p = 0,448).
 Impacto del sacubitrilo-valsartán en la evolución de la clase funcional y B) dosificación del sacubitrilo-valsartán en pacientes hospitalizados en comparación con pacientes ambulatorios. NYHA: New York Heart Association; SV: sacubitrilo-valsartán.")
En los datos analíticos, los pacientes hospitalizados frente a los ambulantes presentan una reducción de la media del filtrado glomerular (MDRD-4) de 69,17 ± 17,12 a 65,67 ± 20,30ml/min y 69,29 ± 28,10 a 68,82 ± 27,35ml/min respectivamente, así como un pequeño aumento no significativo de la creatinina en ambos subgrupos. Existe una reducción de la mediana del NT-proBNP tanto en pacientes hospitalizados como ambulatorios, sin diferencias (3.088 a 563 pg/ml en hospitalizados y 2.335 a 1.150 pg/ml en ambulatorios, p = ns.
La seguridad del fármaco se evaluó en función de la aparición de fracaso renal agudo (FRA) (definido como un aumento de la creatinina > 50% respecto a la basal), hiperpotasemia (potasio sérico > 5,5 mEq/l), hipotensión sintomática o asintomática (PAS < 90mmHg), diarreas, mareos, náuseas y vómitos. En ambos subgrupos no se aprecian diferencias significativas en los efectos secundarios, salvo un pequeño aumento de la incidencia de hiperpotasemia, no significativo y clínicamente no relevante, en pacientes hospitalizados (tabla 1). La hiperpotasemia sucedió en el 11,6 y 11,3% de los pacientes del PIONEER-HF4 y TRANSITION2,3 respectivamente, y en el 16,1% de los pacientes del PARADIGM-HF1. La mayor tasa de hiperpotasemia en nuestro estudio podría estar relacionada con un mayor uso de antagonistas de la aldosterona en nuestra cohorte, respecto al de los ensayos PARADIGM-HF1 y PIONEER-HF4 (54,5 y 10,9% respectivamente, frente a 69,7% en el SAVE-RLIFE).
La discontinuación del SV difiere en ambos grupos, pero no significativamente (13,1 y 6,3%; p = 0,191). Las causas de discontinuación dentro del subgrupo de pacientes ambulatorios fueron: 2,7% por FRA, 2,2% por hipotensión sintomática, 1,6% por falta de respuesta, 1,1% debido al coste del fármaco y el 6% por causas desconocidas. En el subgrupo de pacientes hospitalarios, las causas fueron el 2,1% por FRA y el 4,2% por causas desconocidas. El porcentaje de pacientes fallecidos fue similar (6,6 y 4,2%; p = 0,532) en ambos subgrupos.
El SV se inició tras alcanzar estabilidad clínica y no precisar tratamiento diurético intravenoso. Se comenzó con la dosis baja (24/26mg cada 12 h), excepto en pacientes con una PAS > 110mmHg, un filtrado glomerular estimado > 60ml/min o dosis medias/altas de inhibidores de la enzima de conversión de angiotensina/antagonista del receptor de aldosterona II previamente, en los que se inició a dosis medias (49/51mg cada 12 h). Una vez comenzado el fármaco, se evaluaba al paciente en una primera visita en consulta (entre la segunda y la cuarta semana) y posteriormente en las visitas de seguimiento. Según el estado clínico, la presión arterial y la función renal e iones, se modificaba la dosis tanto del SV como del resto de la medicación para la ICFEr, a criterio del clínico y sin que la dosificación o seguimiento posterior estuvieran estipulados por protocolo. Como se aprecia en la figura 1, en ambos subgrupos se observa una modificación hasta dosis máximas (97/103mg cada 12 h) del 37,5 y 27,9%; p = 0,194 en los subgrupos hospitalizados y ambulatorios respectivamente. Dicha modificación de las dosis es menor a la alcanzada en los ensayos clínicos TRANSITION2,3 (tras la semana 10 de tratamiento del 45 y 50% en el grupo de inicio del SV antes del alta y después del alta respectivamente); y PIONEER-HF4 (55,2% en el grupo del SV a las 8 semanas). Este menor logro de dosis objetivo en nuestro estudio puede deberse a los estrictos protocolos de seguimiento que conllevan los ensayos clínicos comparados con la práctica clínica habitual.
En consistencia con los datos conocidos del ensayo clínico TRANSITION2,3, nuestro estudio en vida real refleja que el SV es un fármaco seguro y eficaz en pacientes con ICFEr y que puede utilizarse tanto de forma ambulatoria como hospitalaria tras una estabilización de un episodio de IC aguda.



