












Cardiología joven: temas de actualidad en salud cardiovascular
More infoLa hipertensión arterial pulmonar (HAP) es una enfermedad rara y grave en la que se produce un remodelado vascular pulmonar anómalo con aumento de las resistencias vasculares pulmonares, presiones pulmonares y poscarga del ventrículo derecho, produciendo insuficiencia cardiaca derecha por fracaso del ventrículo derecho, que es la causa principal de muerte. Avances en el conocimiento de los mecanismos fisiopatológicos de la enfermedad han permitido el desarrollo de nuevos compuestos moleculares dirigidos a revertir al menos parcialmente las alteraciones de la vasculatura pulmonar aportando mecanismos de acción y dianas terapéuticas adicionales a los fármacos vasodilatadores clásicos. Este trabajo presenta una revisión acerca de los mecanismos fisiopatológicos, el proceso diagnóstico de la HAP atendiendo a cómo integrar los diferentes elementos clínicos y pruebas complementarias para un diagnóstico precoz y correcto, y expone los tratamientos disponibles y en desarrollo con análisis del potencial papel de los últimos en los algoritmos terapéuticos actuales.
Pulmonary arterial hypertension (PAH) is a rare and serious pathology in which abnormal pulmonary vascular remodeling occurs causing increased pulmonary vascular resistance, pulmonary pressures and right ventricular afterload, leading to right heart failure due to right ventricular failure, which is the main cause of death. Advances in the understanding of the pathophysiological mechanisms of the disease have allowed the development of new molecular compounds aimed at partially reversing the alterations in the pulmonary vasculature, providing additional mechanisms of action and therapeutic targets to the classic pulmonary vasodilator drugs. This work covers a review of the pathophysiological mechanisms, the diagnostic process of PAH, considering how to integrate the different clinical elements and diagnostic tests for an early and correct diagnosis, and presents the available and developing molecular compounds with an analysis of the potential role of the latter in current therapeutic algorithms.
La hipertensión pulmonar (HP) es un síndrome complejo caracterizado por un remodelado vascular pulmonar obliterativo que produce un aumento de las resistencias vasculares pulmonares (RVP) que generan, en último término, una sobrecarga de presión del ventrículo derecho (VD). Sin tratamiento, el VD acaba claudicando, y la insuficiencia cardiaca es la causa habitual de muerte1. El diagnóstico de certeza se realiza con un cateterismo derecho que determina una presión arterial pulmonar media >20mmHg. Si la presión capilar pulmonar es >15mmHg, la HP es de origen poscapilar. En cambio, si es inferior, hablaremos de HP precapilar. Será HP combinada cuando exista una presión capilar pulmonar elevada y las RVP excedan las 2UW2.
La clasificación actual distingue 5 grupos etiológicos (tablas 1 y 2). Aunque epidemiológicamente el tipo 2 (secundaria a cardiopatía izquierda) es el más frecuente (∼80%), esta revisión se centrará en la hipertensión arterial pulmonar (HAP) (HP del grupo 1) por tener características fisiopatológicas, historia natural y respuesta a los tratamientos bien definidas.
Clasificación clínica de la hipertensión pulmonar y perfil hemodinámico asociado
| Hipertensión arterial pulmonar | HP asociada a cardiopatía izquierda | HP asociada a enfermedadpulmonar | HP asociada a obstrucciones arteriales pulmonares | HP por mecanismos multifactoriales o no conocidos | |
|---|---|---|---|---|---|
| Tipo HPa | HP precapilarb | HP poscapilarHP combinada pre y poscapilar | HP precapilarb | HP precapilarb | HP precapilar o poscapilar |
| PAPm, mmHg | >20 | >20 | >20 | >20 | >20 |
| PCP, mmHg | ≤15 | >15 | ≤15 | ≤15 | ≤15 |
| RVP, UW | >2 | HP poscapilar aislada ≤2HP combinada >2 | >2 | >2 | >2 |
HP: hipertensión pulmonar; PAPm: presión arterial pulmonar media, PCP: presión capilar pulmonar; RVP: resistencias vasculares pulmonares.
Clasificación clínica de la hipertensión pulmonar
| Grupo 1. HAP |
| 1.1. Idiopática |
| 1.1.1. Respondedores a largo plazo a calcioantagonistas |
| 1.2. Hereditariaa |
| 1.3. Asociada con fármacos y toxinasa,b |
| 1.4. Asociada con: |
| 1.4.1. Enfermedad del tejido conectivo |
| 1.4.2. Infección por el VIH |
| 1.4.3. Hipertensión portal |
| 1.4.4. Cardiopatías congénitas |
| 1.4.5. Esquistosomiasis |
| 1.5. HAP con datos de enfermedad venooclusiva/hemangiomatosis capilar pulmonar |
| 1.6. HP persistente del recién nacido |
| Grupo 2. HP secundaria a cardiopatía izquierda |
| 2.1. Debida a insuficiencia cardiaca |
| 2.1.1. Con fracción de eyección preservada |
| 2.1.2. Con fracción de eyección reducida o ligeramente reducidac |
| 2.1.3. Cardiomiopatías con etiologías específicas (hipertrófica, amiloide, Fabry, Chagas) |
| 2.2. Valvulopatías |
| 2.2.1. Valvulopatía aórtica |
| 2.2.2. Valvulopatía mitral |
| 2.2.3. Valvulopatía mixta |
| 2.3. Enfermedades cardiovasculares congénitas/adquiridas que conducen a HP poscapilar |
| Grupo 3. HP secundaria a enfermedades pulmonares o a hipoxia |
| 3.1. Enfermedad pulmonar obstructiva crónica o enfisema |
| 3.2. Enfermedad pulmonar intersticial |
| 3.3. Combinado fibrosis pulmonar y enfisema |
| 3.4. Otra patología parenquimatosa pulmonar (no incluido en grupo 5) |
| 3.5. Patología restrictiva no parenquimatosa (síndromes hipoventilación, neumonectomía) |
| 3.6. Hipoxia sin patología pulmonar (ej. altitud) |
| 3.7. Enfermedades pulmonares del desarrollo |
| Grupo 4. HP asociada con obstrucciones de las arterias pulmonares |
| 4.1. HP tromboembólica crónica |
| 4.2. Otras obstrucciones de las arterias pulmonaresd |
| Grupo 5. HP de mecanismo desconocido o multifactorial |
| 5.1. Enfermedades hematológicase |
| 5.2. Enfermedades sistémicasf |
| 5.3. Enfermedades metabólicasg |
| 5.4. Insuficiencia renal crónica con o sin hemodiálisis |
| 5.5. Microangiopatía trombótica tumoral pulmonar |
| 5.6. Mediastinitis fibrosante |
| 5.7. Cardiopatías congénitas complejas |
HAP: hipertensión arterial pulmonar; HP: hipertensión pulmonar; VIH: virus de inmunodeficiencia humana.
Los pacientes con HAP hereditaria o inducida por fármacos y toxinas pueden ser respondedores a largo plazo a calcioantagonistas.
En la HAP inducida por fármacos y toxinas, estas se clasifican en 2 subgrupos en función de la evidencia científica: «asociación definitiva» y «asociación posible» (tabla 3).
Fracción de eyección ventricular izquierda reducida: ≤40%. Fracción de eyección ventricular izquierda ligeramente reducida: 41-49%.
Otras causas de obstrucción de las arterias bronquiales incluyen: sarcomas (de grado intermedio o alto, o angiosarcoma), otros tumores malignos (carcinoma renal, carcinoma uterino, tumor testicular de células germinales), tumores no malignos (leiomioma uterino), arteritis sin enfermedad del tejido conectivo, estenosis congénita de las arterias pulmonares e hidatidosis.
Incluyendo anemia hemolítica crónica hereditaria o adquirida, y síndromes mieloproliferativos crónicos.
En la HAP se produce un remodelado vascular obliterativo principalmente en las arterias musculares de mediano y pequeño calibre y, en menor medida (salvo en formas concretas como la enfermedad venooclusiva pulmonar), en capilares, vénulas y arteriolas. Entre las alteraciones producidas está la fibrosis intimal y formación de una neoíntima por proliferación descontrolada de células endoteliales, hipertrofia de la media por hiperplasia de células musculares lisas, proliferación de fibroblastos e infiltración de células inflamatorias en la adventicia, neomuscularización de arteriolas, vasoconstricción, trombosis in situ, rarefacción capilar, y formación de lesiones plexiformes en estadios más avanzados1. Finalmente, estas alteraciones repercuten sobre el VD, cuyo remodelado suele ser inicialmente adaptativo, con hipertrofia compensadora y contractilidad aumentada para mantener el volumen sistólico (adaptación homeométrica), pero con el tiempo será maladaptativo, con disminución de la contractilidad y dilatación (adaptación heterométrica) para mantener el volumen sistólico, que finalmente estará disminuido en estadios más avanzados. La disfunción endotelial, del músculo liso vascular y del sistema inflamatorio e inmunitario por diferentes factores genéticos, epigenéticos y ambientales juegan un papel determinante en la HAP, causando una secreción y función alterada de factores de crecimiento, citocinas, hormonas y canales iónicos3. Estos activan las vías de señalización de regulación de los factores de transcripción que, de forma directa y a través de mecanismos epigenéticos, activan genes proliferativos, proinflamatorios y metabólicos4.
A pesar del avance en el conocimiento fisiopatológico de la HAP, aún existen muchas incógnitas sobre las vías moleculares implicadas. Entre las más estudiadas se encuentra la de las proteínas morfogénicas del hueso y las activinas, cuyos receptores pertenecen a la superfamilia de los receptores de los factores de transformación de crecimiento-β, e incluyen al receptor tipo 2 de la proteína morfogénica del hueso3. Las mutaciones de este receptor representan entre el 70-80% de las formas heredables de HAP, además de haberse observado una expresión reducida en los pacientes sin mutaciones5. En la HAP, por tanto, existe una disminución de la señalización a través del receptor tipo 2 de la proteína morfogénica del hueso (antiproliferativo) con un predominio de la señalización a través de receptores proproliferativos de esta superfamilia, que promueven la expresión de genes que desregulan la proliferación, apoptosis o integridad endotelial. Otra de las vías moleculares implicadas es la de los receptores de tirosina cinasa, entre los que se encuentra el receptor del factor de crecimiento derivado de plaquetas α y β cuya expresión y función y la de su ligando, el factor de crecimiento derivado de plaquetas α y β, así como del receptor del factor estimulante de colonias 1 y receptor c-kit están aumentados en la HAP, contribuyendo a la proliferación, fibrosis e inflamación. Por último, se han descrito alteraciones en canales de potasio, factores de transcripción, metabolismo mitocondrial, sistema inmunitario, angiogénesis, activación neurohormonal, estrés oxidativo o matriz extracelular3,4.
DiagnósticoPara el diagnóstico temprano de la HAP, es esencial mantener un alto nivel de sospecha clínica, que debe ser posteriormente confirmado mediante pruebas complementarias.
AnamnesisEl síntoma más temprano asociado a la HAP es la disnea de esfuerzo. Otros síntomas comunes pero que suelen presentarse más tardíamente incluyen la bendopnea, las palpitaciones y la hemoptisis. A medida que avanza la enfermedad, pueden aparecer síntomas de alarma relacionados con fallo del VD y con bajo gasto, como náuseas y distensión gástrica, aumento de peso y síncopes6. Adicionalmente, existen síntomas raros debidos a la dilatación de la arteria pulmonar, tales como el dolor torácico por compresión de la arteria coronaria izquierda, la disfonía por compresión del nervio laríngeo recurrente (síndrome de Ortner), y las atelectasias e infecciones respiratorias secundarias a la compresión bronquial7.
Exploración físicaEn la exploración física destacan los signos típicos de insuficiencia cardiaca derecha, tanto retrógrados (distensión yugular, ascitis, edema periférico) como anterógrados (cianosis, frialdad de extremidades, enlentecimiento del relleno capilar)6. Una exploración física detallada puede ayudar a identificar la causa subyacente de la HAP, ya que existen signos muy característicos de determinadas afecciones (como la cianosis diferencial, sugestiva de ductus arterioso persistente; o las telangiectasias, síndrome de Raynaud y enfermedad por reflujo esofágico, sugestivos de esclerodermia).
Pruebas complementariasElectrocardiogramaPuede observarse desviación derecha del eje, P. pulmonale, signos de hipertrofia ventricular derecha (R/S>1 o R>0,5mm en V1), bloqueo de rama derecha y patrón de sobrecarga del VD (depresión del segmento ST e inversión de la onda T en precordiales derechas) (fig. 1)8.
Radiografía de tórax
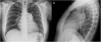
Puede observarse dilatación de cavidades derechas (extensión hacia fuera de la silueta cardiaca en la proyección lateral y contacto del VD con más de un tercio de la longitud del esternón en la proyección posteroanterior) y de la arteria pulmonar, incluyendo aneurisma, así como la pérdida de vasos periféricos (fig. 2)9.

Radiografía de tórax con hallazgos característicos de hipertensión pulmonar.
A) Proyección posteroanterior con dilatación de aurícula derecha (expansión del borde derecho de la silueta cardiaca hacia arriba y afuera) y dilatación de ambas arterias pulmonares. B) Proyección lateral con dilatación del ventrículo derecho (disminución del espacio retroesternal y contacto del ventrículo derecho con >1/3 longitud del esternón).
Independientemente de la causa subyacente, la HAP conlleva una sobrecarga de presión y disfunción progresiva del VD, detectable mediante ecocardiografía10 (fig. 3). El diagnóstico definitivo se obtiene mediante cateterismo derecho, mientras que el ecocardiograma transtorácico solo estima la probabilidad de que exista hipertensión pulmonar. Los hallazgos característicos se resumen en la tabla 3.

Hallazgos ecocardiográficos observados en la hipertensión pulmonar.
A) Plano paraesternal eje corto con septo interventricular aplanado, ventrículo izquierdo en forma de D e índice de excentricidad ventrículo izquierdo >1,1. B) Plano apical 4 cámaras con ventrículo derecho dilatado con cociente basal ventrículo derecho/ventrículo izquierdo >1, aurícula derecha dilatada (>18cm2). C) Plano apical 4 cámaras con insuficiencia tricuspídea severa por técnica Doppler color. D) Plano paraesternal eje corto de grandes vasos con dilatación aneurismática del tronco pulmonar. E) Plano apical 4 cámaras con velocidad de regurgitación tricuspídea>3,4m/s mediante técnica Doppler continuo. F) Plano paraesternal eje corto de grandes vasos con velocidad de regurgitación pulmonar protodiastólica>2,2m/s mediante técnica Doppler continuo. G) Plano paraesternal eje corto de grandes vasos con tiempo de aceleración del tracto de salida del ventrículo derecho <105ms y notch mesosistólico. H) Plano subcostal con derrame pericárdico severo circunferencial.
Características ecocardiográficas sugestivas de hipertensión pulmonar
| Velocidad pico de regurgitación tricuspídea | ||
| Probabilidad baja<2,8m/s | Probabilidad intermedia2,9-3,4m/s | Probabilidad alta>3,4m/s |
| Otros parámetros indirectos sugestivos de hipertensión pulmonar | ||
| Ventrículos | Arteria pulmonar | Vena cava inferior/aurícula derecha |
| • Relación diámetro basal o área VD/VI>1• Aplanamiento septal en sístole o diástole• TAPSE/PAPS <0,55mm/mmHg | • Tiempo de aceleración pulmonar <105ms y/o notch sistólico• Velocidad de regurgitación pulmonar protodiastólica >2,2m/s• Diámetro de arteria pulmonar >25mm o mayor que diámetro raíz aórtica | • Vena cava inferior >21mm con colapso inspiratorio reducido• Área aurícula derecha >18cm2 |
PAPs: presión arterial pulmonar sistólica; TAPSE: excursión sistólica del anillo tricuspídeo; VD: ventrículo derecho; VI: ventrículo izquierdo.
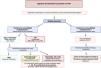
Debido a la inespecificidad inicial de los síntomas, es necesario un alto grado de sospecha clínica para lograr un diagnóstico temprano. En la figura 4 se muestra el algoritmo utilizado en la práctica clínica habitual para la detección de la HAP, que consta de 3 pasos: a) sospecha de HAP en función de anamnesis, exploración física y pruebas complementarias básicas; b) determinación de la probabilidad ecocardiográfica de la HP en aquellos pacientes con alto grado de sospecha clínica, y c) confirmación de la HAP mediante cateterismo derecho en aquellos pacientes con probabilidad ecocardiográfica de la HP intermedia o alta2.

Algoritmo diagnóstico de hipertensión arterial pulmonar. Se muestran los 3 pasos en el diagnóstico de la HAP: sospecha diagnóstica, valoración ecocardiográfica de probabilidad de hipertensión pulmonar y confirmación mediante cateterismo derecho.
aImplica descartar HP del grupo 2 (ecocardiograma transtorácico), grupo 3 (pruebas de función respiratoria, tomografía computerizada pulmonar) y grupo 4 (gammagrafía de ventilación pulmonar).
bAl menos 2 signos pertenecientes a 2 categorías diferentes (tabla 3).
cSi sospecha de hipertensión arterial pulmonar idiopática, hereditaria o asociada a drogas.
dSolicitar serología de virus de inmunodeficiencia humana y virus hepatotropos, perfil de autoinmunidad, ecografía abdominal y estudio genético; valorar estimación cociente flujo pulmonar (Qp)/flujo sistémico (Qs) si sospecha de cardiopatía congénita. HAP: hipertensión arterial pulmonar; NT-proBNP: prohormona N-terminal del péptido natriurético cerebral; O2: oxígeno; VRT: velocidad de regurgitación tricuspídea.
Fuente: Figura creada con BioRender.com.
El tratamiento de los pacientes con HAP es complejo y debe llevarlo a cabo por equipos expertos en unidades especializadas. El abordaje integral incluye medidas generales, tratamientos farmacológicos de soporte y vasodilatadores pulmonares (VDp). Además, dependiendo del subgrupo etiológico y del estadio evolutivo de la enfermedad, se pueden requerir procedimientos intervencionistas o quirúrgicos.
Medidas generalesActividad físicaSe recomienda el entrenamiento físico supervisado en los pacientes estables y con tratamiento farmacológico óptimo, por su impacto positivo en la capacidad de ejercicio y calidad de vida (recomendación IA según últimas guías europeas)2,11.
AnticoagulaciónLa anticoagulación podría considerarse en los pacientes con HAP idiopática en base a resultados de estudios observacionales12, mientras que su uso podría ser potencialmente perjudicial en los pacientes con HAP asociada a conectivopatías, especialmente esclerodermia13. Dada la evidencia contradictoria y la falta de ensayos clínicos aleatorizados, actualmente no se recomienda de forma sistemática. Puede considerarse en los pacientes con aneurisma de la arteria pulmonar >50mm, para prevenir trombosis in situ2.
DiuréticosLa retención de fluido se asocia a peor pronóstico. Los fármacos más utilizados para alcanzar la euvolemia y evitar la congestión residual son los diuréticos de asa, tiazidas y antagonistas de receptores de mineralocorticoides, tanto aislados como en combinación (recomendación IC)2. Mención especial a los últimos, de gran utilidad por contrarrestar la marcada activación del eje renina-angiotensina-aldosterona de los pacientes con insuficiencia cardiaca derecha y que podrían presentar propiedades antifibróticas14.
Oxigenoterapia domiciliaria crónicaA pesar del beneficio fisiopatológico teórico de la oxigenoterapia (produce vasodilatación pulmonar y disminución de las RVP), no existen datos que demuestren el beneficio a largo plazo. Por este motivo, se extrapola la evidencia de la enfermedad pulmonar obstructiva crónica, recomendándose en los pacientes con saturación de oxígeno <92% o presión parcial de oxígeno <60mmHg en al menos 2 ocasiones (recomendación IC)2. Por otro lado, existe evidencia observacional que sugiere una reducción de la mortalidad en los pacientes con disminución grave de la capacidad de difusión del monóxido de carbono (<40% del predicho)15.
Fármacos cardiovascularesNo existen datos acerca del beneficio del uso de inhibidores del receptor de la angiotensina-neprilisina, inhibidores de la enzima convertidora de angiotensina, antagonistas de los receptores de angiotensina II, bloqueadores beta ni inhibidores del cotransportador sodio-glucosa tipo 2, por lo que no se recomienda su utilización sistemática (recomendación IIIC)2.
Anemia y estado del hierroEl déficit de hierro se asocia a un deterioro de la función miocárdica, exacerbación sintomática e incremento de mortalidad. Se define como una ferritina sérica <100 o 100-299μg/l si el índice de saturación de transferrina es <20%. Se recomienda la reposición intravenosa en los pacientes con ferropenia y anemia grave, definida como cifras de hemoglobina <7-8g/dl (nivel de recomendación IC). En ausencia de anemia, podría considerarse, pero con menor nivel de evidencia (IIb)2.
Tratamiento vasodilatador pulmonarEl desarrollo durante las últimas décadas de fármacos específicos para el tratamiento de la HAP, ha cambiado de manera sustancial el abordaje y el pronóstico de esta enfermedad16. La combinación de los diferentes VDp con esquemas de tratamiento cada vez más agresivos han condicionado una importante mejoría en la capacidad de ejercicio, hemodinámica, calidad de vida, tiempo al empeoramiento clínico y supervivencia17,18. En la tabla 4 se recogen los fármacos VDp aprobados para su uso en Europa, su vía de administración, posología y principales efectos adversos.
Vasodilatadores pulmonares
| Fármaco | Vía de administración | Posología | Efectos secundarios |
|---|---|---|---|
| Bloqueadores de los canales de calcio | |||
| Amlodipino | Oral | Inicio 5mg/cada 24hObjetivo 15-30mg/cada 24h | |
| Diltiazem | Oral | Inicio 60mg/cada 12hObjetivo 120-360mg/cada 12h | Cefalea, mareo, hipotensión arterial, edemas, bradicardia |
| Felodipino | Oral | Inicio: 5mg/cada 24hObjetivo: 15-30mg/cada 24h | |
| Nifedipino | Oral | Inicio: 10mg/cada 8hObjetivo: 20-60mg/cada 8-12h | |
| Vía del óxido nítrico | |||
| Inhibidores de fosfodiesterasa-5 (iPDE-5) | |||
| Sildenafilo | Oral | 20-80mg/cada 8h | Cefalea, eritema facial, priapismo, epistaxis, dispepsia. Contraindicados con nitratos o riociguat |
| Tadalafilo | Oral | 40mg/cada 24h | |
| Estimuladores de la guanilato ciclasa soluble (GCs) | |||
| Riociguat | Oral | Inicio: 1mg/cada 8hObjetivo: 2,5mg/cada 8h | Cefalea, dispepsia, mareo, hipotensión. Contraindicado con nitratos o con iPDE5 |
| Vía de la endotelina: antagonistas de los receptores de endotelina (ARE) | |||
| Bosentán | ARE A y BOral | Inicio: 62,5mg/cada 12hObjetivo:125mg/cada 12h | Elevación transaminasas, congestión nasal |
| Ambrisentán | ARE AOral | Inicio: 5mg/cada 24hObjetivo: 10mg/cada 24h | Edema periférico, elevación transaminasas, congestión nasal |
| Macitentán | ARE A y BOral | 10mg/cada 24h | Anemia, congestión nasal |
| Vía de las prostaciclinas | |||
| Agonistas de los receptores IP | |||
| Selexipag | Oral | Inicio: 200μg/cada 12hObjetivo: 1.600μg/cada 12h | Trombocitopenia, eritema facial, cefalea, dolor mandibular, náuseas/vómitos, diarreaEpoprostenol problemas relacionados con bomba de infusión intravenosa continuaTreprostinil subcutáneo dolor local |
| Prostanoides | |||
| Epoprostenol | Intravenoso | Inicio: 2ng/kg/minObjetivo: 20-40ng/kg/min | |
| Treprostinil | Intravenoso,subcutáneo | Dosis inicial: 4ng/kg/minObjetivo: 40-80ng/kg/min | |
| Iloprost | Inhalado | Inicio: 2,5μg/inh/cada 4hObjetivo: 5μg/inh/cada 4h | |
IP: receptor I de la prostaciclina.
Los primeros ensayos clínicos con fármacos VDp realizados en los pacientes con nuevo diagnóstico de HAP se basaban en un solo fármaco, eran de corta duración, con una población relativamente pequeña y enfocados a valorar cambios en la capacidad funcional, medida habitualmente mediante la prueba de marcha de 6min (PM6M)19–21. Sin embargo, en la última década, los ensayos clínicos han evolucionado hacia diseños más ambiciosos, con poblaciones mayores y distintos esquemas de terapias combinadas (secuenciales inicialmente y más recientemente combinados de inicio)18,22–24. Además, emplean objetivos primarios más robustos, basados en el deterioro clínico y la supervivencia a medio/largo plazo25–27. La tabla 5 recoge los principales ensayos clínicos con fármacos VDp aprobados en Europa.
Principales ensayos clínicos con vasodilatadores pulmonares en hipertensión arterial pulmonar
| Estudio | Molécula | Sujetos (n) | Duración | Objetivo primario | Resultado | Comentario |
|---|---|---|---|---|---|---|
| Epoprostenol19 | Epoprostenol i.v. | 81 | 12sem | Cambio distancia recorrida PM6M | Positivo | PM6M ↑32m grupo de epoprostenol iv + terapia convencional (348±17msem 12 frente a 316±18m basal) y ↓15m grupo de terapia convencional (257± 24msem 12 frente a 272±23m basal); p<0,003 |
| BREATHE-135 | Bosentán | 213 | 16sem | Cambio distancia recorrida PM6M | Positivo | PM6M ↑36m grupo bosentán y ↓8m grupo placebo (diferencia media 44m (IC 95%: 21-67; p<0,001) |
| EARLY36 | Bosentán | 185 | 6 meses | Cambio en las RVP y cambio distancia recorrida PM6M | Positivo | Mejoría RVP grupo bosentán frente a placebo (−22,6% [IC 95%: −33,5;−10,0; p<0,001]); ↑ PM6M bosentán frente a placebo (19,1m [IC 95%: 3,6-41,8; p=0,0758]) |
| ARIES-134 | Ambrisentán | 202 | 12sem | Cambio distancia recorrida PM6M | Positivo | Mejoría grupos ambrisentán frente a placebo: ↑31m (IC 95%: 3-59; p=0,008) ambrisentán 5mg y 51m (IC 95%: 27-76; p<0,001) ambrisentán 10mg |
| ARIES-234 | Ambrisentán | 192 | 12sem | Cambio distancia recorrida PM6M | Positivo | Mejoría grupos ambrisentán frente a placebo: 32m (IC 95%: 2-63; p=0,022) ambrisentán 2,5mg y 59m (IC 95%: 30-89; p<0,001) ambrisentán 5mg |
| SUPER-129 | Sildenafilo | 277 | 12sem | Cambio distancia recorrida PM6M | Positivo | ↑PM6M en grupos tratamiento sildenafilo frente a placebo: 45m dosis 20mg (IC 99%: 21-70; p<0,001); 46m dosis 40mg (IC 99%: 20-72; p<0,001); 50m dosis 80mg (IC 99%: 23-77; p<0,001) |
| PACES30 | Sildenafilo | 267 | 16sem | Cambio distancia recorrida PM6M | Positivo | PM6M ↑29,8m en grupo sildenafilo y 1m en grupo placebo: diferencia media 28,8m (IC 95%: 13,9-43,8m; p<0,001) |
| PHIRST31 | Tadalafilo | 405 | 16sem | Cambio distancia recorrida PM6M | Positivo | ↑PM6M en grupo tadalafilo 10, 20, 40mg (no 2,5mg) frente a placebo.Solo dosis 40mg alcanzó significación estadística (p<0,01): efecto medio tratamiento 33m (IC 95%: 15-50m) |
| PATENT-132 | Riociguat | 443 | 12sem | Cambio distancia recorrida PM6M | Positivo | PM6M ↑30m en el grupo riociguat 2,5mg y ↓6m en grupo placebo: diferencia media 36m (IC 95%: 20-52; p<0,001) |
| REPLACE33 | Riociguat | 226 | 24sem | Mejoría: no empeoramiento clínico + mejoría ≥2 de: PM6M, CF OMS, NT-proBNP | Positivo | El objetivo primario se consigue en 45 (41%) de los 111 pacientes en grupo riociguat y en 23 (20%) de los 113 pacientes grupo IPDE5; OR: 2,78 (IC 95%: 1,53-5,06; p=0,0007) |
| AIR38 | Iloprost inh | 203 | 12sem | Combinado: ↑ ≥10% PM6M + ↑CF NYHA + no deterioro clínico o muerte | Positivo | Efecto positivo del tratamiento a favor de iloprost frente a placebo; OR: 3,97 (IC 95%: 1,47-10,75; p=0,007) |
| STEP39 | Iloprost inh | 67 | 12sem | PM6M, CF NYHA, parámetros hemodinámicos y tiempo hasta el deterioro clínico | Positivo | ↑PM6M en grupo iloprost frente a placebo: efecto medio tratamiento 26m (p=0,051). CF ↑34 frente al 6% (p=0,002). Iloprost retrasa tiempo a deterioro clínico (p=0,0219).↑hemodinámica PAPm (−8mmHg; p<0,001) y RVP (−254dyn · s · cm-5; p<0,001) |
| Treprostinil sc20 | Treprostinil s.c. | 470 | 12sem | Cambio distancia recorrida PM6M | Positivo | PM6M ↑10m grupo treprostinil frente a 0m en grupo placebo: diferencia media entre grupos 16m (IC 95%: 4,4-27,6; p=0,006) |
| TRIUMPH23 | Treprostinil inh | 235 | 12sem | Cambio distancia recorrida PM6M | Positivo | PM6M ↑21,6m en grupo treprostinil, 3m en grupo placebo: diferencia media 20m (IC 95%: 8-32,8m; p[eCMH]=0,0004; p[WRS]=0,0016) |
| FREEDOM-M39 | Treprostinil oral | 349 | 12sem | Cambio distancia recorrida PM6M | Positivo | Efecto positivo tratamiento treprostinil frente a placebo: efecto medio del tratamiento 23m (IC 95%: 4-41m; p=0.0125) |
| FREEDOM-C24 | Treprostinil oral | 350 | 16sem | Cambio distancia recorrida PM6M | Negativo | ↑PM6M no alcanzó significación estadística: efecto medio tratamiento 11m (IC 95%: −2-22; p=0,089) |
| FREEDOM-C221 | Treprostinil oral | 310 | 16sem | Cambio distancia recorrida PM6M | Negativo | ↑PM6M no alcanzó significación estadística: efecto medio tratamiento 10m (p=0,07) |
| SERAPHIN37 | Macitentán | 742 | Duración media: 85,3sem (placebo), 99,5sem (macitentán 3mg), 103,9sem (macitentán 10mg) | Tiempo inicio tratamiento hasta 1.° evento clínico: muerte, septostomía auricular, trasplante pulmonar, inicio prostaciclinas iv/sc, progresión HAP | Positivo | Macitentán reduce significativamente riesgo evento morbimortalidad:- Macitentán 3mg frente a placebo HR: 0,70 (IC 97,5%: 0,52-0,96; p=0,01)- Macitentán 10mg frente a placebo HR: 0,55 (IC 97,5%: 0,39-0,76; p<0,001) |
| AMBITION18 | Ambrisentán Tadalafilo | 500 | Duración media estudio 517 días (550 días grupo tratamiento combinado, 484 días grupo monoterapia, p=0,03) | Tiempo inicio tratamiento hasta 1er deterioro clínico: muerte, hospitalización empeoramiento HAP, progresión enfermedad, o respuesta clínica insatisfactoria | Positivo | La combinación inicial de ambrisentán + tadalafilo disminuye el riesgo de sufrir un evento de deterioro clínico frente a al tratamiento en monoterapia con ambrisentán o tadalafilo: HR: 0,50 (IC 95%: 0,35-0,72; p<0,001) |
| COMPASS-222 | BosentánSildenafilo | 334 | Seguimiento 39,7±22,6 meses para el grupo placebo y 38,0±21,9 meses para el grupo bosentán | Tiempo desde inicio del tratamiento hasta primer evento de morbimortalidad | Negativo | Bosentán+ sildenafilo no superior a monoterapia sildenafilo para retrasar 1.° evento morbimortalidad: evento morbimortalidad 51,4% placebo y 42,8% bosentán (HR: 0,83; IC 97,31%: 0,58-1,19; p=0,2508) |
| GRIPHON25 | Selexipag | 1156 | Duración media del estudio 63,7sem (placebo) y 70,7sem (selexipag) | Tiempo inicio tratamiento hasta 1.° evento combinado muerte o complicación HAP | Positivo | Selexipag ↓ significativamente riesgo de sufrir evento morbimortalidad frente a placebo: HR: 0,60 (IC 99%: 0,46-0,78; p<0,001) |
| TRITON26 | TadalafiloMacitentán Selexipag | 247 | 26sem | Cambio en las RVP | Negativo | Triple terapia combinada inicio (tadalafilo+macitentán+selexipag) y doble terapia combinada de inicio (tadalafilo+macitentán) ↓RVP (54 y 52%, respectivamente), sin diferencias significativas entre grupos (efecto tratamiento: 0,96; IC 95%: 0,86-1,07; p=0,42). |
CF: clase funcional; eCMH: Cochran-Mantel-Haenszel ampliado; HAP: hipertensión arterial pulmonar; HR: hazard ratio; IC 95%: intervalo de confianza del 95%; inh: inhalado; iv: intravenoso; NT-ProBNP: fracción aminoterminal del propéptido natriurético cerebral tipo B; NYHA: New York Heart Association; OR: odds ratio; PAPm: presión arterial pulmonar media; PM6M: prueba de marcha de 6 min; RVP: resistencias vasculares pulmonares; sc: subcutáneo; sem: semana; WRS: suma de rangos de Wilcoxon.
Los antagonistas de los canales del calcio están indicados en los pacientes con respuesta positiva a la prueba vasodilatadora realizada durante el cateterismo cardiaco derecho. Existe evidencia de su beneficio en la HAP idiopática, hereditaria e inducida por drogas/toxinas. Los fármacos utilizados son el amlodipino, diltiazem, nifedipino o felodipino. Se administran en monoterapia por vía oral. Se deben iniciar a dosis bajas con incremento progresivo vigilando posibles efectos adversos: hipotensión, edema periférico y bradicardia.
Aproximadamente la mitad de los pacientes con respuesta aguda positiva la mantiene a largo plazo, por lo que debe evaluarse la respuesta clínica y hemodinámica a los 3-6 meses de alcanzar la dosis objetivo. Se considerará respondedor cuando mantiene una situación de bajo riesgo con valores hemodinámicos normalizados o casi (idealmente presión arterial pulmonar media ≤30mmHg y RVP≤4UW)28. Si la respuesta no es satisfactoria, o los pacientes respondedores a largo plazo pierden la respuesta, deben añadirse fármacos VDp específicos sin retirar el antagonista de los canales del calcio, ya que esto puede condicionar un deterioro clínico2.
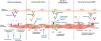
Vasodilatadores pulmonares específicosLos vasodilatadores pulmonares están indicados en los pacientes con HAP y test de vasorreactividad negativo o en los pacientes con test de vasorreactividad positivo y falta de respuesta a largo plazo al tratamiento con antagonistas de los canales del calcio. Los fármacos aprobados se clasifican en 3 grupos en función de la vía fisiopatológica sobre la que actúan (fig. 5)1.

Vías implicadas en la fisiopatología de la hipertensión arterial pulmonar y mecanismo de acción de los fármacos. AC: adenilato ciclasa; ActRIIA/B: receptor de activina tipo IIA/B; ALK: cinasa similar a los receptores de activina; AT: adenosín trifosfato; AMPc: adenosín monofosfato cíclíco; BMP: proteína morfogénica del hueso; BMPRII: receptor tipo 2 de la proteína morfogénica del hueso; ET A: receptor de endotelina A; ET B: receptor de la endotelina B; GCs: estimulante de la guanilato ciclasa; GDFs: factores de diferenciación del crecimiento; GMP: guanosín monofosfato; GMPc: guanosín monofosfato cíclico; GTP: guanosín trifosfato; PDE5: fosfodiesterasa 5.
Fuente: Figura creada con BioRender.com.
Los pacientes con HAP presentan una disminución de la producción y biodisponibilidad del óxido nítrico en el endotelio vascular pulmonar. El óxido nítrico estimula a la guanilato ciclasa soluble, que a su vez cataliza la formación de guanosín monofosfato cíclico, que induce relajación del músculo liso vascular. El óxido nítrico es, además, inhibidor de la proliferación celular en el músculo liso y de la activación plaquetaria.
Los fármacos de la vía del óxido nítrico se clasifican según su mecanismo de acción en inhibidores de la fosfodiesterasa-5 (iPDE-5) o estimulantes de la guanilato ciclasa soluble. Sildenafilo y tadalafilo son inhibidores competitivos reversibles de la PDE-5, responsable de la degradación del guanosín monofosfato cíclico29–31. Riociguat es un estimulante de la guanilato ciclasa soluble, que la estabiliza en configuración activa en situaciones de baja disponibilidad o ausencia de óxido nítrico32,33.
Vía de la endotelinaLa endotelina-1, producida fundamentalmente en las células del endotelio vascular, es un potente vasoconstrictor y estimulador de la proliferación celular, que ejerce sus efectos sobre los receptores ETA y ETB. Los niveles pulmonares y circulantes de endotelina 1 están aumentados en los pacientes con HAP. La vía de la endotelina 1 puede bloquearse con los fármacos antagonistas de los receptores de la endotelina selectivos (ambrisentán)34 o no selectivos (bosentán y macitentán)35–37.
Vía de las prostaciclinasLas prostaciclinas endógenas se sintetizan en las células endoteliales, se unen preferencialmente al receptor I de la prostaciclina activando el adenosín monofosfato cíclico. Tienen un efecto vasodilatador, antitrombótico, antiinflamatorio y antiproliferativo sobre la pared de los vasos. Los pacientes con HAP tienen niveles reducidos debido a la menor expresión de prostaciclina sintasa en las arterias pulmonares. Las terapias que actúan a través de esta vía son los análogos de la prostaciclina (epoprostenol, iloprost y treprostinil)19,20,30,38–40 y el agonista selectivo del receptor IP de la prostaciclina (selexipag)25.
Nuevos fármacos antirremodeladoCon el avance en el conocimiento de las vías moleculares implicadas en la HAP se han desarrollado nuevos compuestos moleculares dirigidos directamente contra estas, para revertir, al menos parcialmente, el remodelado vascular pulmonar anómalo. Se están desarrollando compuestos dirigidos contra los distintos mecanismos patogénicos en diferente situación de desarrollo3,4.
SotaterceptRepresenta el fármaco antirremodelado con un desarrollo más avanzado. Actúa en las vías de las activinas/proteínas morfogénicas del hueso que están mediadas a través de la superfamilia de los receptores de factores de transformación de crecimiento-β. Los ligandos proteínas morfogénicas del hueso se unen al receptor tipo 2 de la proteína morfogénica del hueso (receptor de factores de transformación de crecimiento-β tipo 2) que heterodimeriza con la cinasa similar a los receptores de activina 1/2/3/6 (receptor de factores de transformación de crecimiento-β tipo 1) activándose una cascada de señalización de genes anti-proliferativos. Las activinas y los factores de diferenciación del crecimiento se unen al heterodímero de la cinasa similar a los receptores de activina ALK 4/5/7 y al receptor de activina tipo IIA/B (receptor de factores de transformación de crecimiento-β tipo 2) activando genes proproliferativos. En la HAP existe un predominio de las vías proproliferativas. El sotatercept es una proteína de fusión compuesta por el dominio extracelular del receptor de activina tipo IIA unido al dominio Fc de la IgG1 humana que actúa atrapando ligandos proproliferativos (activinas y factores de diferenciación del crecimiento), restaurando parcialmente el equilibrio entre las vías anti y proproliferativas4 (fig. 5). Se administra subcutáneamente cada 21 días. En modelos preclínicos ha demostrado revertir el remodelado vascular pulmonar y ventricular derecho y disminuir la inflamación. En el ensayo clínico fase 2 PULSAR de los pacientes con HAP en clase funcional II/III altamente tratados (35% doble terapia, 56% triple, 37% infusión de prostaciclina) se alcanzó el objetivo primario de reducción de las RVP y los objetivos secundarios de empeoramiento clínico, mejoría de la fracción aminoterminal del propéptido natriurético cerebral tipo b (NT-proBNP) y la PM6M41, los cuales se mantuvieron en la fase abierta del ensayo a 18-24 meses42. Cabe destacar que la mejoría de las RVP se produjo a expensas de una reducción de presiones pulmonares, sin modificación del gasto cardiaco ni de la presión capilar pulmonar, sugiriendo un efecto directo sobre el remodelado vascular. El ensayo clínico fase 3 STELLAR incluyó a una población de HAP de largo tiempo de evolución (media de tiempo desde diagnóstico 9 años) altamente tratados (61% triple terapia, 40% infusión de prostaciclinas). Se alcanzó el objetivo primario de aumento de la distancia de la PM6M, así como 8/9 objetivos secundarios clínicos, hemodinámicos, funcionales y analíticos, incluyendo una reducción clínicamente muy relevante del 84% del tiempo hasta la muerte o empeoramiento clínico y que ocurrió muy precozmente (∼10 semanas) y se mantuvo en el tiempo43. Además, un análisis post hoc del efecto hemodinámico y de función cardiaca derecha, mostró una mejoría significativa de presiones pulmonares, sin cambios en el gasto cardiaco, mejorando la compliancia (rigidez) y elastancia (poscarga) arterial pulmonar, el acoplamiento del VD-arteria pulmonar y las dimensiones y función del VD44. Actualmente está en desarrollo la fase abierta de ambos ensayos clínicos, el estudio SOTERIA (NCT04796337) que pretende evaluar la tolerabilidad, eficacia y seguridad largo plazo. El sotatercept es un fármaco bien tolerado, con pocos efectos adversos graves. Entre los más frecuentes están la poliglobulia, por su efecto eritropoyético, la trombocitopenia idiosincrásica, los sangrados, en su mayoría leves, y las telangiectasias. El sotatercept se aprobó para el tratamiento de la HAP en EE. UU. (03.2024) y acaba de serlo en Europa (08.2024); se espera que se apruebe por la Agencia Española del Medicamento próximamente.
Inhibidores de los receptores de tirosina cinasaEl primer inhibidor de los receptores de tirosina cinasa que se desarrolló para la HAP fue el imatinib oral. En el ensayo clínico fase 3 IMPRES imatinib mostró una mejoría en la distancia de la PM6M, así como hemodinámica45. Sin embargo, la mala tolerancia y el desarrollo de efectos adversos frecuentes y graves incluyendo hematoma subdural en 8 pacientes anticoagulados dio lugar a que no se permitiera su uso en HAP46. Para evitar los efectos adversos sistémicos se desarrolló una formulación inhalada que fue investigada en un ensayo clínico fase 2b/3 (IMPAHCT, NCT05036135), con resultados neutros que llevaron a su finalización.
El seralutinib es un potente inhibidor de varios receptores de tirosina cinasa diseñado específicamente para el tratamiento de la HAP de administración inhalada. Recientemente se han publicado los resultados del ensayo clínico fase 2 donde se evaluaba su eficacia y seguridad frente a placebo en los pacientes con HAP en clase funcional II/III y RVP>5UW y que incluyó una población con gravedad hemodinámica (RVP medias 8,4UW) pero buena situación clínica global (68% clase funcional II; NT-proBNP medio 188ng/l; mediana de distancia PM6M 409m), observando una reducción estadísticamente significativa del objetivo primario de las RVP (1,2UW) a expensas de descenso de presiones pulmonares sin modificación de gasto cardiaco, mayor en los pacientes con peor clase funcional y mayor gravedad hemodinámica. El fármaco fue bien tolerado, y el efecto adverso más frecuente fue la tos47.
OtrosAdemás del sotatercept, en la vía de señalización de las proteínas morfogénicas del hueso/activinas se están desarrollando otros compuestos moleculares. Actualmente está en marcha un ensayo clínico fase 2 con el compuesto KER-012, una proteína de fusión con estructura similar a la del sotatercept que atrapa activinas y, por tanto, disminuye las vías de señalización proproliferativas de los receptores del factor de transformación de crecimiento-β (NCT05975905). Otra potencial diana terapéutica es el BMP9, ligando del receptor tipo 2 de la proteína morfogénica del hueso, cuya función es compleja y con evidencia preclínica contradictoria, sugiriéndose que si su receptor es wildtype el BMP9 activa vías de señalización antiproliferativas, pero si está mutado es proproliferativo48.
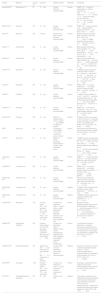
Algoritmo terapéuticoEn el momento de iniciar el tratamiento VDp hay que realizar una estratificación de riesgo multiparamétrica para evaluar si el paciente se encuentra en riesgo bajo, intermedio o alto (fig. 6), además de tener en cuenta otros aspectos como la comorbilidad o el acceso al tratamiento2. En los pacientes sin comorbilidad cardiopulmonar en riesgo bajo o intermedio al diagnóstico se recomienda doble terapia oral de inicio con iPDE-5+antagonistas de los receptores de la endotelina (recomendación I según últimas guías europeas) tras los resultados del ensayo AMBITION donde se demostró la superioridad de la doble terapia oral de inicio frente a monoterapia18. No se recomienda la triple terapia oral de inicio tras los resultados del ensayo TRITON, que no mostró un beneficio adicional a la doble terapia26. En los pacientes en alto riesgo al diagnóstico se recomienda triple terapia de inicio con iPDE-5+antagonista de receptor de endotelina más análogo de la prostaciclina sc/iv, acorde a evidencia de vida real por ser la combinación más potente (recomendación IIa)2,17,19,20,49. En los pacientes en riesgo intermedio al diagnóstico, pero con gravedad hemodinámica puede plantearse triple terapia de inicio17. Tras el comienzo del tratamiento y en el seguimiento deberá realizarse una reevaluación periódica del riesgo con clasificación en 4 estratos (riesgo bajo, intermedio/bajo, intermedio/alto y alto; fig. 6) para establecer la necesidad de escalar el tratamiento si el paciente no está en bajo riesgo (salvo excepciones)2,50. En riesgo intermedio/bajo con doble terapia oral se añadirá selexipag (recomendación IIa) de acuerdo con los resultados del ensayo GRIPHON, donde se observó beneficio de este compuesto añadido al tratamiento con iPDE-5 o antagonistas de los receptores de la endotelina; alternativamente se cambiará el iPDE-5 por riociguat (recomendación IIa) por los resultados del ensayo abierto REPLACE, donde se observó una mejoría clínica con este cambio25,33. Si el paciente está en riesgo intermedio-alto o alto con tratamiento oral habrá que añadir análogo de la prostaciclina sc/iv o iniciar evaluación para trasplante pulmonar (recomendación IIa)2. Actualmente, con la aparición del inhibidor de la señalización de las activinas, sotatercept, se abre una cuarta vía terapéutica con un fármaco antirremodelado. Aunque todavía existen incógnitas respecto al posicionamiento exacto del fármaco, se podrá plantear en el seguimiento de los pacientes con HAP que se encuentren fuera del bajo riesgo añadido a cualquier combinación de fármacos VDp51.

Estratificación del riesgo en hipertensión arterial pulmonar, adaptado de las guías de la Sociedad Europea de Cardiología de práctica clínica de 2022. BNP: péptido natriurético de tipo B; FEVD: fracción de eyección del ventrículo derecho; IC: índice cardiaco; iSV: índice volumen sistólico; iVTSVD: índice de volumen telesistólico ventricular derecho; NT-proBNP: fracción N-terminal del propéptido natriurético tipo B; OMS: Organización Mundial de la Salud; PAD: presión auricular derecha; PAPs: presión arterial pulmonar sistólica; SvO2: saturación de oxígeno venosa mixta; TAPSE: desplazamiento sistólico del plano del anillo tricuspídeo; VE/VCO2: equivalente ventilatorio de dióxido de carbono; VO2: consumo de oxígeno.
Recientemente, en los registros de la HAP se ha observado un cambio epidemiológico con diagnóstico más tardío (∼6.ª década) y con asociación frecuente de comorbilidades que impacta el pronóstico y tratamiento. No existe consenso acerca de cómo definir y cuantificar la comorbilidad cardiopulmonar; como comorbilidad cardiovascular se incluyen enfermedades que predisponen a la disfunción diastólica ventricular izquierda (hipertensión arterial, diabetes mellitus, obesidad, fibrilación auricular, cardiopatía isquémica...), y como pulmonar enfermedades parenquimatosas pulmonares, que suelen asociar capacidad de difusión de monóxido de carbono reducida <45% del predicho. En los pacientes comórbidos la tolerancia y el cumplimiento terapéutico son peores, desarrollan más efectos adversos y la evidencia científica es limitada por haber sido frecuentemente excluidos de los ensayos clínicos52. Existe más experiencia con iPDE-5, que parecen ser los fármacos con mejor tolerancia y perfil de seguridad, siendo los antagonistas de los receptores de la endotelina los de más riesgo de retención hídrica en presencia de disfunción diastólica izquierda. En los pacientes con comorbilidad pulmonar, el tratamiento VDp puede empeorar la oxigenación por empeoramiento de la relación ventilación/perfusión53. Por ello, en los pacientes con HAP con comorbilidad se recomienda en la mayoría monoterapia oral de inicio (recomendación IIa), preferiblemente con iPDE-5, aunque en casos de alto riesgo puede plantearse un tratamiento inicial más agresivo (p. ej., doble terapia oral secuencial) en un entorno hospitalario, para controlar la tolerancia2. Aunque la evaluación en el seguimiento en 4 estratos de riesgo es más limitada por la falta de evidencia del tratamiento, puede servir de orientación para plantear una escalada secuencial de tratamiento individualizada si el paciente está en una situación subóptima de riesgo, siempre teniendo en cuenta los efectos adversos, el techo terapéutico y que en este grupo alcanzar el riesgo bajo puede no ser un objetivo realista54. Por último, resulta clave tener un control óptimo de las comorbilidades y la situación de volemia con ajuste de diuréticos antes del inicio del tratamiento VDp para minimizar el riesgo de efectos adversos2,51. En la figura 7 se presenta el algoritmo terapéutico al diagnóstico y en el seguimiento.

Algoritmo terapéutico en la hipertensión arterial pulmonar. Para valoración del riesgo inicial (3 estratos) y en el seguimiento (4 estratos) ver figura 6.
aNo existe consenso acerca de cómo definir y cuantificar la comorbilidad cardiopulmonar.
bEn las últimas guías europeas existe un nivel de recomendación IIa añadir a la doble terapia oral un ARPC (selexipag), y IIb para sustituir iPDE-5 por sGC (riociguat).
cEn algunos casos en alto riesgo al diagnóstico puede plantearse un tratamiento más agresivo, p. ej., doble terapia oral iPDE-5+ARE en entorno hospitalario. dEn los pacientes con comorbilidad cardiopulmonar la evidencia del tratamiento según situación de riesgo es más limitada.
APC: agonista de la prostaciclina (epoprostenol i.v.; treprostinil s.c.); ARE: antagonista del receptor de la endotelina (ambrisentán, bosentán, macitentán); ARPC: agonista del receptor de la prostaciclina (selexipag); iPDE-5: inhibidor de la fosfodiesterasa 5 (sildenafilo, tadalafilo); HAP: hipertensión arterial pulmonar; sGC: estimulante de la guanilato ciclasa (riociguat).
Fuente: Figura creada con BioRender.com.
El desarrollo de los nuevos fármacos antirremodelado supone un cambio de paradigma en el tratamiento de la HAP. Los fármacos utilizados hasta la fecha presentan un efecto predominante VDp y por ello la mejoría hemodinámica de disminución de las RVP y de poscarga VD se produce a expensas principalmente de un aumento del gasto cardiaco con poco descenso de las presiones pulmonares. En contraposición, los nuevos fármacos antirremodelado, producen una mejoría hemodinámica por disminución de las presiones pulmonares sin aumentar el gasto cardiaco, lo que se interpreta como un efecto directo antiproliferativo, sin efecto VDp. El sotatercept constituye actualmente una cuarta vía de tratamiento para los pacientes con HAP que proporciona un beneficio adicional a simplemente la estabilización o el enlentecimiento de la enfermedad: pretende revertir, al menos parcialmente, el remodelado vascular pulmonar patológico. Se espera un amplio uso del fármaco y que esto impacte significativamente en la supervivencia de la HAP, una enfermedad que actualmente continúa siendo grave, con pronóstico sombrío, e incurable51. Sin embargo, existen aún diferentes cuestiones por resolver en torno a este compuesto. En primer lugar, el posicionamiento definitivo del mismo pues, aunque potencialmente puede utilizarse con cualquier combinación de fármacos vasodilatadores, hay que recordar que los pacientes incluidos en los ensayos PULSAR y STELLAR presentaban un largo tiempo de evolución de la enfermedad y estaban altamente tratados41–43. Por ello existen actualmente otros ensayos clínicos en curso para aportar evidencia en contextos clínicos concretos de la HAP: HYPERION (NCT04811092) en la HAP de reciente diagnóstico, ZENITH (NCT04896008) en HAP en techo terapéutico como terapia de rescate, CADENCE (NCT04945460) en la HP poscapilar combinada con RVP>4UW. Todo ello, además de la experiencia a largo plazo en la fase abierta de los ensayos y en vida real, aportará más evidencia para poder concretar el posicionamiento definitivo del fármaco en el algoritmo terapéutico, además de su seguridad a largo plazo. Existen otros elementos relevantes como es su precio de comercialización que puede ser determinante según el país y su sistema nacional de salud, así como otras características como la vía de administración y su preparación, subcutánea y en entorno hospitalario/supervisado. Adicionalmente, el fármaco está contraindicado en los pacientes con hemoglobina por encima del límite superior de la normalidad por su efecto eritropoyético. Por ello, el desarrollo de nuevos fármacos con mecanismo de acción también en la vía de las activinas/proteínas morfogénicas del hueso pueden representar alternativas o tratamiento adicional al sotatercept. Además, queda por conocer si la vía inhalada mediante el inhibidor del receptor de tirosina cinasa seralutinib se consolidará como otro nuevo tratamiento antirremodelado, si podrá añadirse al arsenal terapéutico de la HAP y aportará sinergia con sotatercept, planteando más de 4 vías de tratamiento. Queda también por esclarecer la eficacia de estos compuestos en los pacientes comórbidos. Por último, a más largo plazo es esperable que la genética y epigenética jueguen un papel en el desarrollo de tratamientos más individualizados3–5.
Adicionalmente al reto terapéutico se plantea un reto de pruebas diagnósticas y pronósticas que evalúen directamente el efecto de los nuevos fármacos antirremodelado, de hecho, no existe una definición acerca de qué significa «remodelado reverso» o «modificación de la enfermedad», y con las pruebas actuales observamos datos indirectos hemodinámicos, clínicos, de imagen, analíticos o funcionales, y la realización de biopsias presenta un riesgo prohibitivo.
ConclusionesLa HAP constituye una enfermedad rara, grave y sin tratamiento curativo. A lo largo de las últimas décadas se ha producido un cambio sustancial en la atención terapéutica, con un abordaje más agresivo y con tratamientos combinados desde el inicio para frenar la progresión de la enfermedad. Actualmente nos encontramos en una era de cambio de paradigma en el tratamiento de esta enfermedad, acercándonos al tratamiento de su origen fisiopatológico, con el objetivo de revertir el daño en la vasculatura pulmonar y cambiar su curso. Resulta clave continuar avanzando en el conocimiento de las complejas vías de señalización y las interacciones entre las mismas que determinan las alteraciones fisiopatológicas de la HAP para poder comprender qué combinación de compuestos moleculares podrá revertirlas y con ello modificar la historia natural de la HAP.
FinanciaciónI. Martín de Miguel es receptora de un Contrato Río Hortega del ISCIII (CM23/00235). M. Rivas-Lasarte es receptora de un Contrato Juan Rodés del ISCIII (JR20/00003).
Declaración sobre el uso de inteligencia artificialNo se ha utilizado inteligencia artificial.
Contribución de los autoresTodos los autores han participado en la concepción y en el diseño del manuscrito, en la redacción de su borrador, en la revisión del contenido intelectual y en la aprobación definitiva de la versión presentada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



