










El síndrome de deleción 22q11.2 (SD 22q11.2), también conocido como síndrome de DiGeorge, fenotípicamente se ha asociado a cardiopatías conotruncales. El objetivo de esta revisión fue evaluar el impacto del SD 22q11.2 en la mortalidad de los pacientes pediátricos con defectos conotruncales corregidos.
MétodosLa búsqueda bibliográfica se realizó en las bases de datos PubMed, ScienceDirect, Google Scholar y Redalyc. Abarcó los últimos 10 años. Se incluyeron artículos originales sobre mortalidad en población pediátrica con SD 22q11.2 y defectos cardiacos conotruncales corregidos. Se realizó un análisis estadístico de 5 trabajos. Se evaluó la odds ratio (OR), los intervalos de confianza del 95% (IC95%) y la significación estadística, utilizando la prueba χ2 para cada variable de exposición. Para medir el riesgo de sesgo se utilizó el instrumento ROBINS-E.
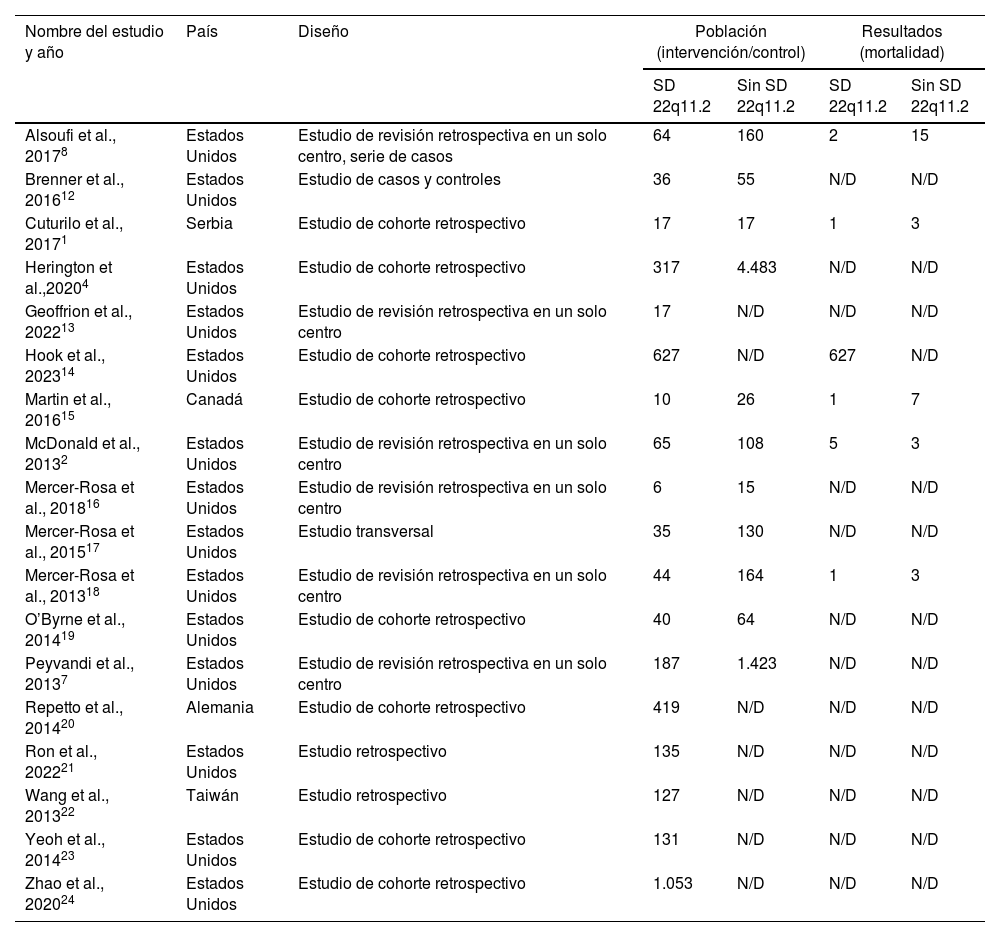
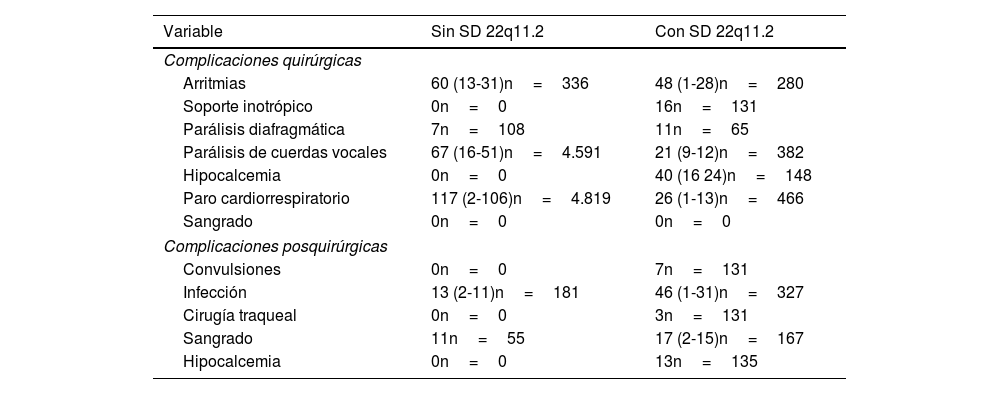
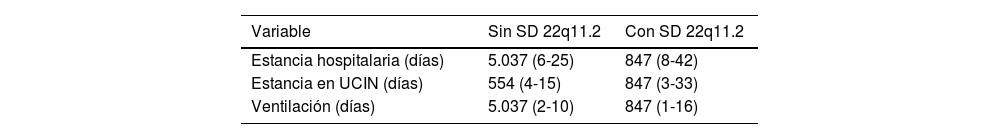
ResultadosSe recabaron 18 artículos, con una población total de 10.993 pacientes (3.225 con SD 22q11.2). La cardiopatía más frecuente fue la tetralogía de Fallot (TF) (n=655). Las complicaciones intraoperatorias más frecuente fueron las arritmias; y las posquirúrgicas fueron las infecciones. Se evaluó la mortalidad en pacientes con SD 22q11.2. La OR fue 0,74 (IC95%, 0,38-1,45; p=0,38). La prueba de heterogeneidad arrojó una I2=66%.
ConclusionesEl SD 22q11.2 no se asocia a la mortalidad posquirúrgica en los pacientes pediátricos con defectos conotruncales.
The 22q11.2 deletion syndrome (22q11.2 DS), known as DiGeorge syndrome, is phenotypically associated with conotruncal heart defects. The aim of this review was to evaluate mortality in pediatric patients with corrected conotruncal defects.
MethodsA literature search was conducted in the databases PubMed, ScienceDirect, Google Scholar, and Redalyc databases, covering the last 10 years. Original articles with pediatric population diagnosed with 22q11.2 DS and conotruncal congenital heart defects were included. A statistical analysis of 5 studies was performed, evaluating the odds ratio (OR), 95% confidence intervals (95%CI), and statistical significance using the chi-square test for each exposure variable. The ROBINS-E tool was used to assess the risk of bias.
ResultsA total of 18 articles were collected, including a total population of 10993 patients (3225 with 22q11.2 DS). The most frequent cardiac defect was tetralogy of Fallot (n=655). The most common intraoperative complications were arrhythmias, and the most common postoperative complications were infections. Mortality in patients with 22q11.2 DS was assessed. The OR was 0.74 (95%CI, 0.38-1.45; P=.38). Heterogeneity test yielded an I2=66%.
Conclusions22q11.2 DS is not associated with post-surgical mortality in patients treated for conotruncal congenital heart defects.
Article
Use datos de acceso a SEC en el menú Acceder.
Si es socio de la Sociedad Española de Cardiología y no puede acceder con sus claves, escriba a rec@cardioclinics.org.
Use the Society's website login and password here.
If you are member of SEC and you have some problems with your login data, please contact with rec@cardioclinics.org.




